N. Tessa Pierce-Ward 0000-0002-2942-5331
· bluegenes
· saltyscientist
Department of Population Health and Reproduction, University of California, Davis
· Funded by NSF 1711984
Luiz Irber 0000-0003-4371-9659
· luizirber
· luizirber
Graduate Group in Computer Science, UC Davis; Department of Population Health and Reproduction, University of California, Davis
Erich M. Schwarz 0000-0003-3151-4381
· SchwarzEM
Department of Molecular Biology and Genetics, Cornell University
C. Titus Brown 0000-0001-6001-2677
· ctb
Department of Population Health and Reproduction, University of California, Davis
Abstract
Metagenomics has expanded our knowledge of microbial diversity, but contaminant sequences are frequently accidentally included in metagenome-assembled genomes.
Genome contamination is often estimated by the presence of marker genes that are biased against detecting contaminants lacking these sequences.
Further, most contamination detection tools do not remove contamination.
We present charcoal, a tool that rapidly identifies and removes contamination in metagenome-assembled genomes using k-mer based methods.
K-mers are nucleotide sequences of length k.
Sufficiently long k-mers are usually specific to a taxonomic lineage.
Taking advantage of this property of k-mers, charcoal identifies majority and minority lineages for each contiguous sequence in a genome and removes contiguous sequences belonging to minority lineages when those lineages occur below a taxonomic threshold (by default, order).
Applying charcoal to the Genome Taxonomy Database rs207, we found approximately XX% of genomes in GTDB were contaminated, with contamination broadly distributed across species and occurring in representative and RefSeq genomes.
Genomes with longer contiguous sequences were less likely to be contaminated.
Our results show concordance with CheckM on detecting the presence of contamination in a genome.
Charcoal is a snakemake workflow developed around the tool sourmash.
It is available at github.com/dib-lab/charcoal, and is pip installable.
Introduction
Metagenomic sequencing has expanded our knowledge of microbial communities and their diversity [1,2,3].
De novo metagenome analysis has generated thousands of draft genomes, termed metagenome-assembled genomes (MAGs), from organisms from diverse environments [2,4,5,6].
Recently, large-scale re-analysis efforts have led to a rapid expansion in draft genomes in public repositories like the European Nucleotide Archive [6] and the Joint Genome Institute IMG/M [2].
Increased observation of draft genomes across the tree of life better enables researchers to contextualize new sequencing data and the roles that microorganisms play in diverse metabolic processes [7,8].
MAG inference relies on assembly and binning of metagenomic sequencing data.
Assembly produces long contiguous sequences by identifying overlaps between short sequencing reads, while binning groups assembled sequences into MAGs using read coverage and tetramernucleotide frequency.
Both processes are subject to biases that can reduce the completeness of or increase the contamination in a MAG: low sequencing coverage or high genomic variation causes short read assemblers to break contiguous sequences into shorter pieces (CITE), which decreases the signal for and accuracy of binning (CITE).
Commonly, the completeness and purity of MAGs is estimated through the presence and sequence composition of single-copy marker genes [9][10], with MAGs that reach >90% completeness and <5% contamination considered high quality [9].
Single-copy marker genes are sets of genes that are present once in a genome of almost all members of a taxonomic group [9,11].
Using these genes to estimate contamination leads to two important biases.
First, given the assumption that marker genes are universally present in genomes, if a marker gene resides on a contaminant sequence but no other sequence for that marker gene is present in the genome, it will not be detected as contamination [9,12].
When a MAG is substantially complete, this may lead to a small underestimation in contamination (~3% [9]), but as completeness decreases, contamination may be substantially underestimated [12].
Second, contiguous sequences which do not contain marker sequences are not included among contamination estimates [9] (note checkm called out plasmids and phages for this bias specifically).
Given these biases, methods that do not rely solely on marker genes may be better suited for contamination estimation.
Many tools have recently been developed that rely on different strategies for detecting contamination [13].
GUNC expands beyond marker genes and uses the full complement of genes in a genome to identify contiguous sequences that fall outside of the predicted lineage [14].
It estimates the distribution of taxonomic assignments within and across contiguous sequences to identify contaminants even among sequences that are poorly taxonomically labelled [14].
Conterminator detects cross-kingdom contamination using all-vs-all alignment and is geared toward contamination detection in large collections of genomes (e.g. databases) [15].
Sequences are considered contaminants when at least 100 nucleotides and no more than 20 kb share a sequence identity of at least 90% in genomes that originate from different kingdoms [15].
In addition, conterminator finds contamination in protein sequences by identifying protein clusters that contain sequences from cross-kingdom members [15].
Removing contamination is a separate problem from estimating its extent.
Tools like RefineM [7], MAGPpurify [16], and BlobTools [17] rely on a combination of GC content, tetramernucleotide frequency, read coverage, phylogenetic or clade markers, conspecific sequences, and known contaminants to flag contaminant contiguous sequences for removal.
The inclusion of read coverage profiles necessitates the use of the original sequencing reads to produce coverage profiles in BAM format for the genomes.
Given that sequencing data and BAM files are usually orders of magnitude larger than genome sequences, this requirement limits the utility of such approaches, especially for large-scale genome databases.
Tools like GUNC are poised for contamination removal without relying on read coverage profiles, but as of yet such approaches are unimplemented [14].
Long k-mers capture relatedness between organisms, where a k=31 captures species-level similarity [18].
K-mers offer an alternative metric to identify contamination, especially in sequences lacking marker genes.
Here we describe Charcoal, an automated method for filtering contaminant contiguous sequences from MAGs.
We show…
We show…
We envisage that charcoal will complement marker gene-based approaches for contamination estimation, removing problematic sequences before they are further analyzed or propagated in public databases.
Methods
Overview
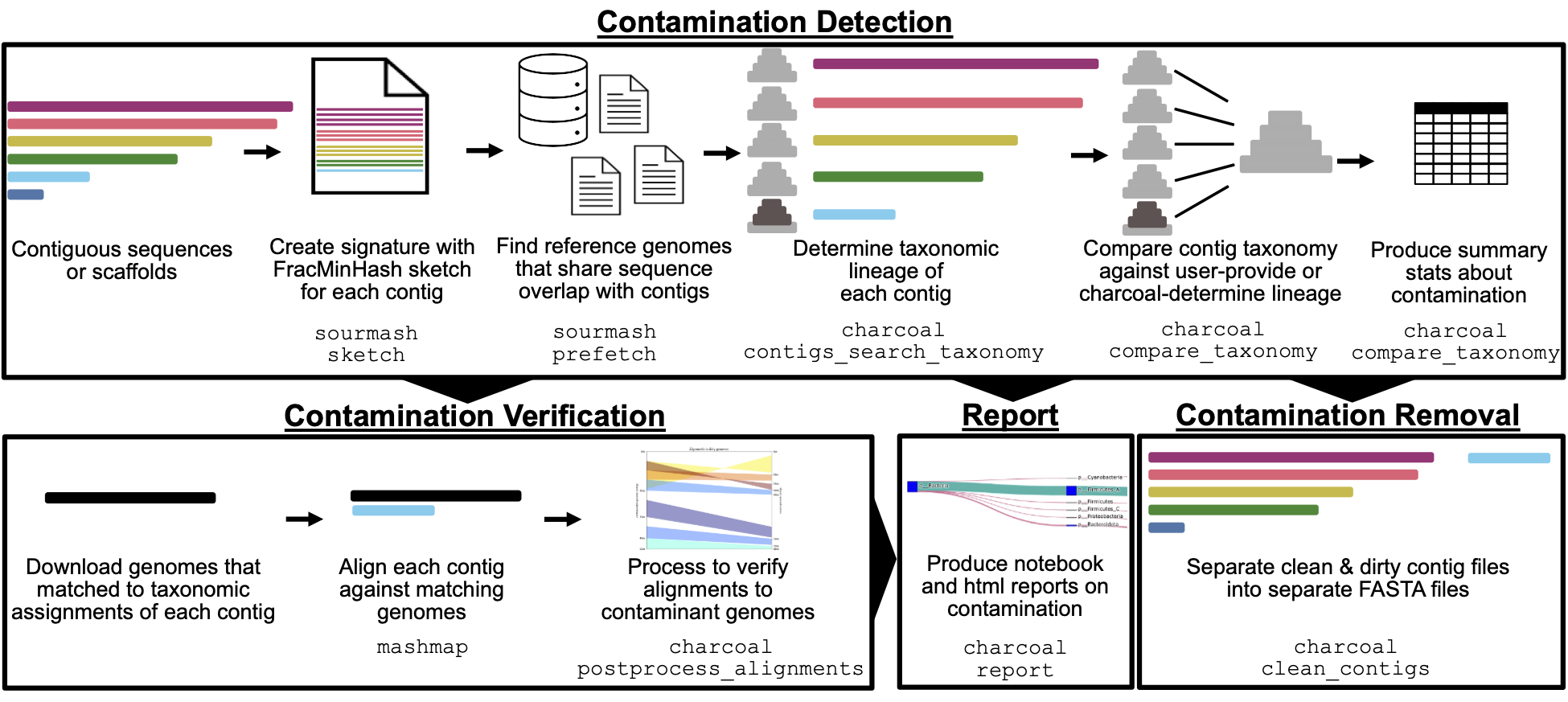
Figure 1:Summary of steps used to decontaminate genomes with charcoal.
In the first stage of the pipeline, contamination detection occurs by comparing the taxonomic assignment of each contig in a genome against all other contigs.
Contigs with inconsistent taxonomy are flagged as contaminants, by default if they differ at the order level or above.
The results of this step are summarized to report the contaminating genomes, the number of contaminant contigs and base pairs at each level of taxonomy, and other metrics like the fraction of the genome that was identifiable and the fraction that was assignable to the majority lineage.
The outputs of the first stage of the pipeline can then be used in any of three additional reporting steps.
First, contamination can be verified by aligning the contaminant contigs against their identified matching reference genome, and the mappings can be visualized.
Second, an html document can be produced that gives an overview of the contamination in each genome.
Lastly, the contaminant contigs can be separated out from the non-contaminant contigs, writing two separate FASTA files per genome.
Charcoal provides an automated method to detect, visualize, and remove bacterial and archaeal contamination in genomes (Figure 1).
To identify contamination, charcoal first creates a FracMinHash sketch for each contiguous sequence (“contig”) in an input genome.
Charcoal then identifies all genomes in a database (“reference genomes”) that share sequence overlap with the input genome using sourmash prefetch.
Subsequent operations subset the original database to include only the genomes identified by prefetch, reducing search volumes.
Using these matches, charcoal uses sourmash gather to identify the minimum set of genomes that cover (or contain) the k-mers in each contig [19].
Charcoal then determines the taxonomic lineage of each contig using a lineage spreadsheet that records the taxonomy of each reference genome in the database; the taxonomic assignment occurs at the lowest common ancestor of all taxonomic assignments given to a contig.
If there is an exact match between the input genome and a genome in the database, this match is removed to allow decontamination to continue.
Charcoal compares the taxonomic lineage of each contig against the lineage of the input genome.
If the contig has a different lineage before or at the configured taxonomic rank (order by default) than that of the majority lineage, the contig is considered a contaminant.
For each contig, charcoal reports the fraction of identified hashes (total and to each major and minor lineage), an estimate of contaminated base pairs (at each taxonomic rank match), as well as the fraction of contigs and base pairs unable to be identified.
The input genome lineage can be user-provided, or it can be determined by charcoal via majority vote of all lineages assigned to all contigs.
If the lineage is determined by charcoal, by default a minimum 10% of the input genome must have been assigned a taxonomic lineage, and 20% of assigned sequences must match to the majority lineage.
If these specifications are not met, charcoal will not decontaminate the input genome unless the user specifies a lineage.
The user can optionally specify a lineage (e.g. d__Eukaryota), and charcoal will remove contigs that have a lineage different from the user-specified one.
This allows charcoal to remove contigs from an input genome when some contigs from that genome occur in the database and when the input genome is not related to anything in a database.
Charcoal reports whether the provided lineage agrees with k-mer classification at or above the genus level.
After the initial stage of contaminant detection, charcoal can perform additional tasks to verify, summarize, or remove the contaminant sequences.
Contaminant verification downloads the reference genome sequence for any genome that was detected among the input genome contigs and aligns the contigs against those genomes using mashmap [20].
Contaminant removal separates “clean” from “dirty” contigs and outputs each set into a FASTA file.
Importantly, charcoal will not remove a contig if it is unidentifiable, whether it is too short to be sketched or does not contain sequences in the reference database.
While these contigs could still be contaminants, charcoal assumes contigs are clean for which it has no information.
Therefore, charcoal will fail to detect contamination for very short contigs which contain no selected k-mers, as well as contigs with novel DNA content.
Lastly, charcoal has a report feature that summarizes and visualizes the taxonomic lineages detected in each input genome as well as the alignments between the input genome and reference genomes.
Sourmash prefetch is the most compute intensive step in the decontamination process.
When using the GTDB rs207 representative database, this step does not exceed 8GB of RAM.
statement that charcoal is database dependent (probably can go in last paragraph with RAM/CPU considerations)
Availability and dependencies
Charcoal is written in python3 and can be installed via pip as charcoal-bio.
The core algorithms (contaminant detection and removal) depend on sourmash and snakemake.
Contaminant verification depends on lxml and mashmap [20].
Reporting depends on mummer, papermill, notebook, and plotly [21].
The source code is available at github.com/dib-lab/charcoal.
ZENODO DOI.
Datasets and benchmarking
We first use charcoal to decontaminate GTDB rs207.
Results
Charcoal detected and removed contamination from roughly one quarter of GTDB genomes
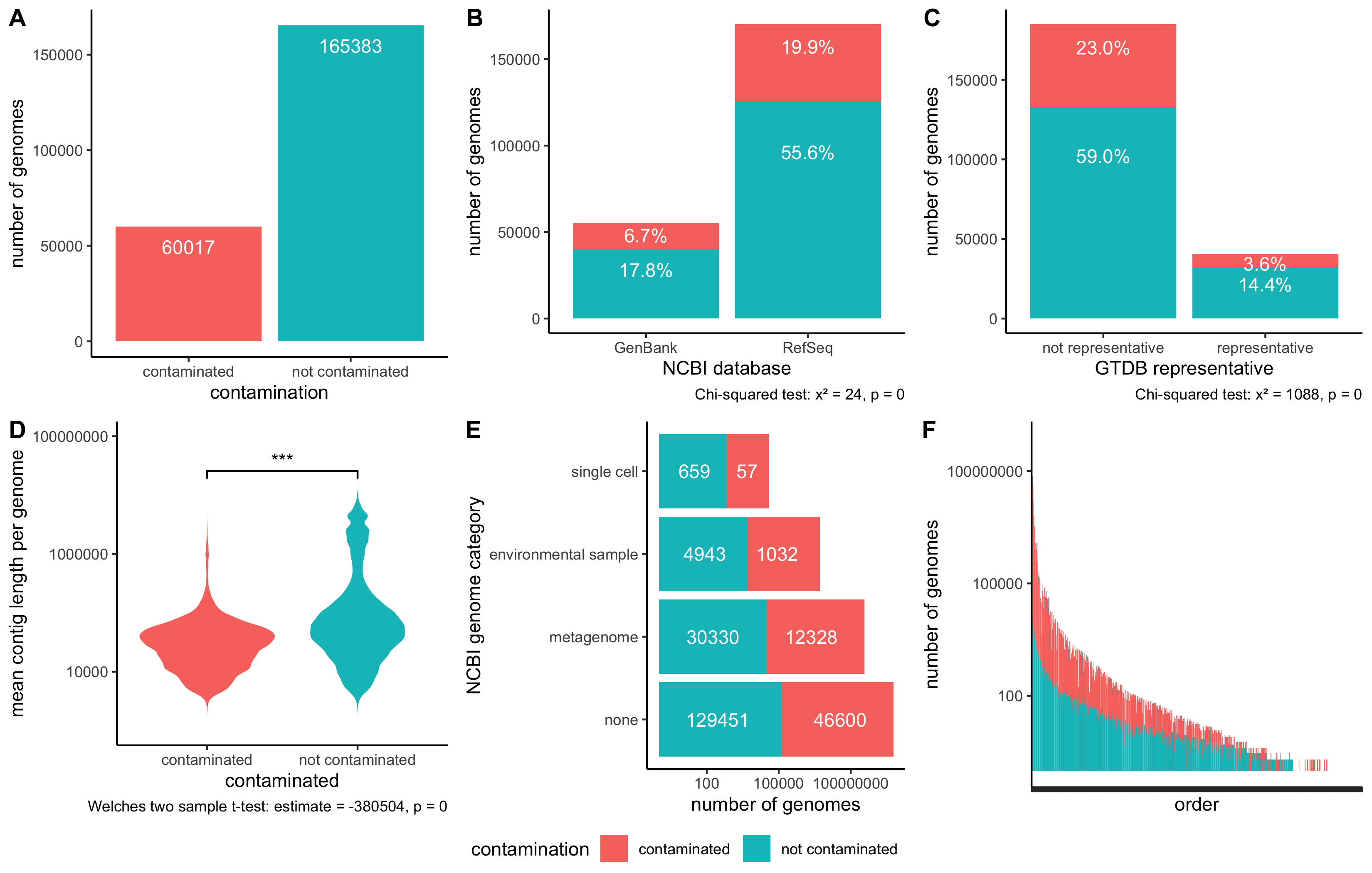
Figure 2:Charcoal identified contamination in approximately one quarter of GTDB rs207 genomes, distributed generally across different types of genomes.A) Bar plot depicting the number of genomes in GTDB contaminated at the order level.
B) Bar plot separating genomes in GTDB by their inclusion in NCBI’s RefSeq. Both GenBank and RefSeq genomes are contaminated.
C) Bar plot separating representative genomes in GTDB from non-representative genomes. While some representative genomes are contaminated, non-representative genomes are more likely to be contaminated.
D) Violin plots depicting the mean contig length within genomes in GTDB, separated by contamination profile. Genomes with longer contigs are less likely to be contaminated. On average, the contigs in contaminated genomes were 380,504 base pairs shorter than contigs in non-contaminated genomes.
E) Bar plot that separates genomes by NCBI category which specifies where source material was derived from.
F) Bar plot depicting the number of genomes in each order, colored by contamination status. Genomes are grouped by taxonomic lineage up to the order level.
Using charcoal to detect and remove contamination, approximately 26% of GTDB rs207 genomes were contaminated at the order level or above (Figure 2 A).
Contamination was distributed across GenBank and RefSeq genomes (Figure 2 B), GTDB representative and non-representative genomes (Figure 2 C), isolate, single cell, and metagenome-derived genomes (Figure 2 E), and taxonomic orders (Figure 2 F).
However, contamination was more likely to be identified in genomes with shorter contigs (Figure 2 D) and in non-representative GTDB genomes (Figure 2 C).
TODO: run prodigal/bakta on dirty contigs, comment on number of genes/average functional potential added to contaminated genomes by contamination.
Charcoal and CheckM identify the same set of not contaminated genomes
Charcoal is more conservative than CheckM contamination estimation at the order level.
whereas 80.2% of genomes are estimated to have some contamination using CheckM.
+ Both tools agree on the majority of genomes with no contamination
At the family level, charcoal identified contamination in XX% of genomes, suggesting that adjusting taxonomy may improve agreement with checkm.
Do plot – at what % checkm contam best overlaps with order level charcoal?
run checkm on clean
Databases
gtdb rs207 reps vs gtdb rs202 reps
gtdb rs202 reps vs gtdb rs202 full (replace evnetually with rs207)
short contigs missed
ram considerations
future development: species-level database
old results outline:
charcoal vs. checkm
charcoal vs. checkm: mgnify, tara
prokka on charcoal dirty
charcoal vs. refineM and magpurify
verification of contamination/contam case studies
case studies
user-provided lineages
cDBG/spacegraphcats?
user-specified lineages
Discussion
Benefits and drawbacks of charcoal.
Commonly, the completeness and contamination of MAGs is estimated through the presence and sequence composition of single-copy marker genes [9,10], with MAGs that reach >90% completeness and <5% contamination considered high quality [9].
Using these genes to estimate contamination leads to two important biases.
First, given the assumption that marker genes are universally present in genomes, if a marker gene resides on a contaminant sequence but no other sequence for that marker gene is present in the genome, it will not be detected as contamination [9,12].
When a MAG is substantially complete, this may lead to a small underestimation in contamination (~3% [9]), but as completeness decreases, contamination may be substantially underestimated [12].
Second, contiguous sequences which do not contain marker sequences are not included among contamination estimates [9].
Charcoal avoids these pitfalls by using k-mers, which are sampled evenly across the genome [19].
We envisage charcoal acting as a complementary analysis tool to CheckM.
While charcoal avoids the above biases, it is itself biased against detecting contamination in very short contigs.
By default, a minimum of three k-mers must be taxonomically annotated on a contig for a contig to be removed; because contigs are first sketched, many short contigs will not contain three k-mers.
Future work
strain heterogeneity estimation or strain insertion in newick tree using prefetch & clean results
completeness estimation, potentially via comparison to conspecifics via genome size
database with eukaryotic sequences
estimation via protein sequences for divergent organisms not well represented in databases
Notes
fastani is an accepted way to do average nucleotide identity calculate, and it relies on k-mers.
from checkm paper:
> Bias in genome quality estimates: Quality estimates based on individual marker genes or collocated marker sets exhibit a bias resulting in completeness being overestimated and contamination being underestimated. This bias is the result of marker genes residing on foreign DNA that are otherwise absent in a genome being mistakenly interpreted as an indication of increased completeness as opposed to contamination.
we don’t deal with strain heterogeneity, as this occurs below the species-level aggregation in the LCA
References
1.
A new view of the tree of life
Laura A Hug, Brett J Baker, Karthik Anantharaman, Christopher T Brown, Alexander J Probst, Cindy J Castelle, Cristina N Butterfield, Alex W Hernsdorf, Yuki Amano, Kotaro Ise, … Jillian F Banfield
Stephen Nayfach, Simon Roux, Rekha Seshadri, Daniel Udwary, Neha Varghese, Frederik Schulz, Dongying Wu, David Paez-Espino, I-Min Chen, Marcel Huntemann, …
Metagenomic compendium of 189,680 DNA viruses from the human gut microbiome
Stephen Nayfach, David Páez-Espino, Lee Call, Soo Jen Low, Hila Sberro, Natalia N Ivanova, Amy D Proal, Michael A Fischbach, Ami S Bhatt, Philip Hugenholtz, Nikos C Kyrpides
Community structure and metabolism through reconstruction of microbial genomes from the environment
Gene W Tyson, Jarrod Chapman, Philip Hugenholtz, Eric E Allen, Rachna J Ram, Paul M Richardson, Victor V Solovyev, Edward M Rubin, Daniel S Rokhsar, Jillian F Banfield
Extensive Unexplored Human Microbiome Diversity Revealed by Over 150,000 Genomes from Metagenomes Spanning Age, Geography, and Lifestyle
Edoardo Pasolli, Francesco Asnicar, Serena Manara, Moreno Zolfo, Nicolai Karcher, Federica Armanini, Francesco Beghini, Paolo Manghi, Adrian Tett, Paolo Ghensi, … Nicola Segata
A unified catalog of 204,938 reference genomes from the human gut microbiome
Alexandre Almeida, Stephen Nayfach, Miguel Boland, Francesco Strozzi, Martin Beracochea, Zhou Jason Shi, Katherine S Pollard, Ekaterina Sakharova, Donovan H Parks, Philip Hugenholtz, … Robert D Finn
proGenomes2: an improved database for accurate and consistent habitat, taxonomic and functional annotations of prokaryotic genomes
Daniel R Mende, Ivica Letunic, Oleksandr M Maistrenko, Thomas SB Schmidt, Alessio Milanese, Lucas Paoli, Ana Hernández-Plaza, Askarbek N Orakov, Sofia K Forslund, Shinichi Sunagawa, … Peer Bork
Rokubacteria: Genomic Giants among the Uncultured Bacterial Phyla
Eric D Becraft, Tanja Woyke, Jessica Jarett, Natalia Ivanova, Filipa Godoy-Vitorino, Nicole Poulton, Julia M Brown, Joseph Brown, MCY Lau, Tullis Onstott, … Ramunas Stepanauskas